
[ad_1]
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) consists of four major structural proteins, of which include the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins. The importance of the S protein in recognizing and interacting with the angiotensin-converting enzyme 2 (ACE2) receptor on the surface of human cells is well known, and the influence of temperature on the normal functioning and structure of this protein has been reported widely.
 Study: How does temperature affect the dynamics of SARS-CoV-2 M proteins? Insights from Molecular Dynamics Simulations. Image Credit: 3dfine / Shutterstock.com
Study: How does temperature affect the dynamics of SARS-CoV-2 M proteins? Insights from Molecular Dynamics Simulations. Image Credit: 3dfine / Shutterstock.com
Viral membrane proteins are the most abundant of the structural proteins in SARS-CoV-2. Beyond merely coating the virus, these proteins are heavily involved in viral assembly and propagation.
In a study recently published on the preprint server bioRxiv*, the structural dynamics of the SARS-CoV-2 M protein are studied at temperatures of 10-50 ˚C using molecular dynamics simulations. To this end, the researchers found that M protein activity peaks at relevant body temperatures and a surprising return to low-temperature behavior beyond this.
Study findings
The M proteins of viruses within the coronavirus family bear high levels of structural and functional similarity, with an outwardly exposed amino-terminal and nucleocapsid-facing carboxy-terminal domain. M proteins must be able to associate with copies of themselves through a link near the transmembrane region, as well as the N, S, and E proteins at the carboxy-terminal domain.
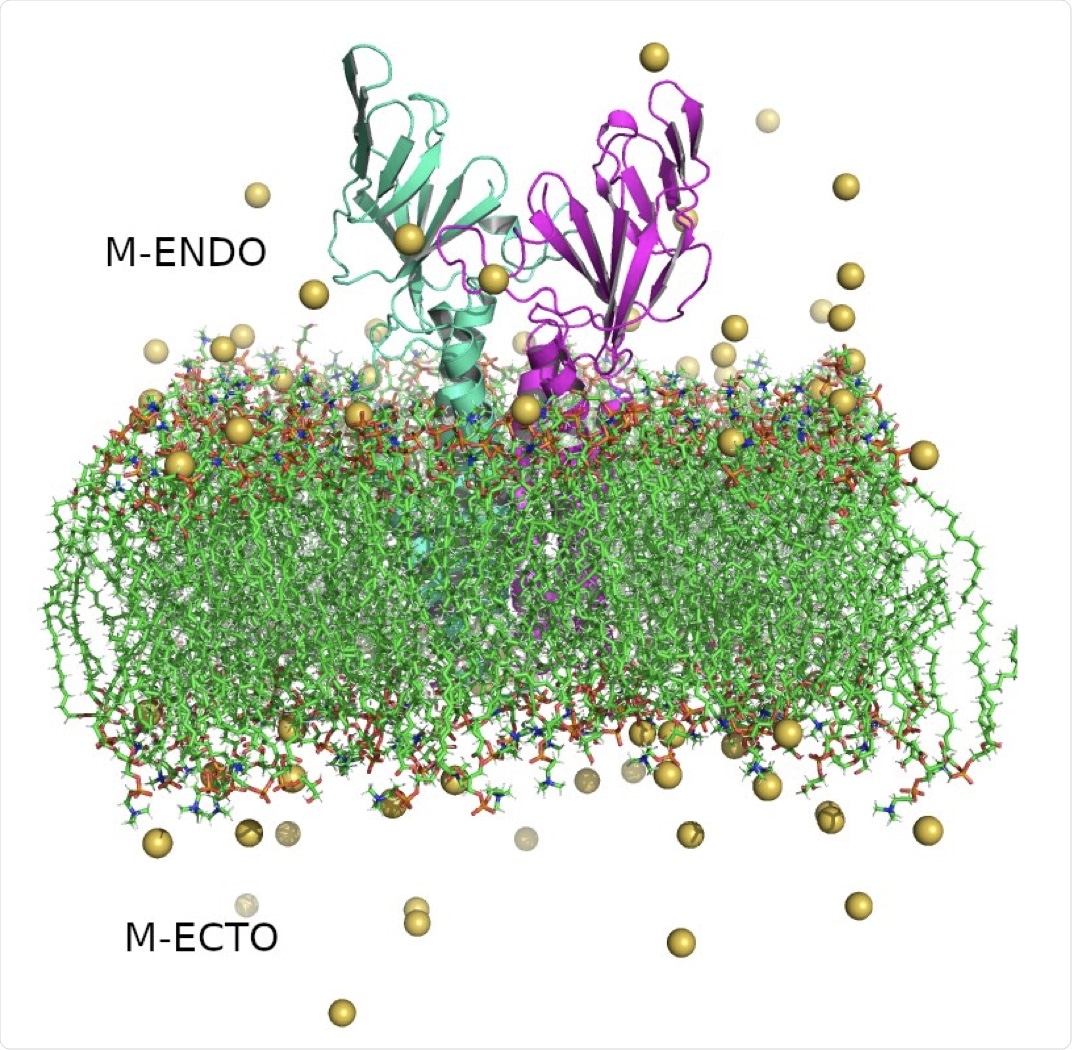 Structure of SARS-CoV-2 Membrane protein dimer. Monomers are shown in cyan and magenta in cartoon representation, lipid bilayers in CPK representation, and neutralizing sodium ions are shown as yellow vdW spheres.
Structure of SARS-CoV-2 Membrane protein dimer. Monomers are shown in cyan and magenta in cartoon representation, lipid bilayers in CPK representation, and neutralizing sodium ions are shown as yellow vdW spheres.
The researchers of the current study obtained a lowest-energy conformer model for the M protein that had been shown by previous simulations to be stable in a lipid bilayer, a single molecule of which was subsequently embedded in a lipid bilayer of dipalmitoylphosphatidylcholine (DPPC) and dipalmitoylphosphoglycerol (DPPG) lipids in a 7:3 ratio. Water molecules, as well as sodium and chloride ions, were added to the system, which constituted a 10 x 10 x 13 nanometer (nm) box.
The root mean square of deviation is a measure of the conformational flexibility of a protein and was assessed at temperatures of 10˚C, 20˚C, 30˚C, 40˚C, and 50˚C in 150 nanoseconds (ns) long simulations. The system was found to be relatively stable at each temperature and was somewhat higher at elevated temperatures.
An area of the protein in the outer part of the transmembrane helix near the C-terminal was noted to change positions at temperatures of 40 ˚C and 50 ˚C, with several other solvent-exposed residues close to the N-terminal domain also undergoing notable shifts.
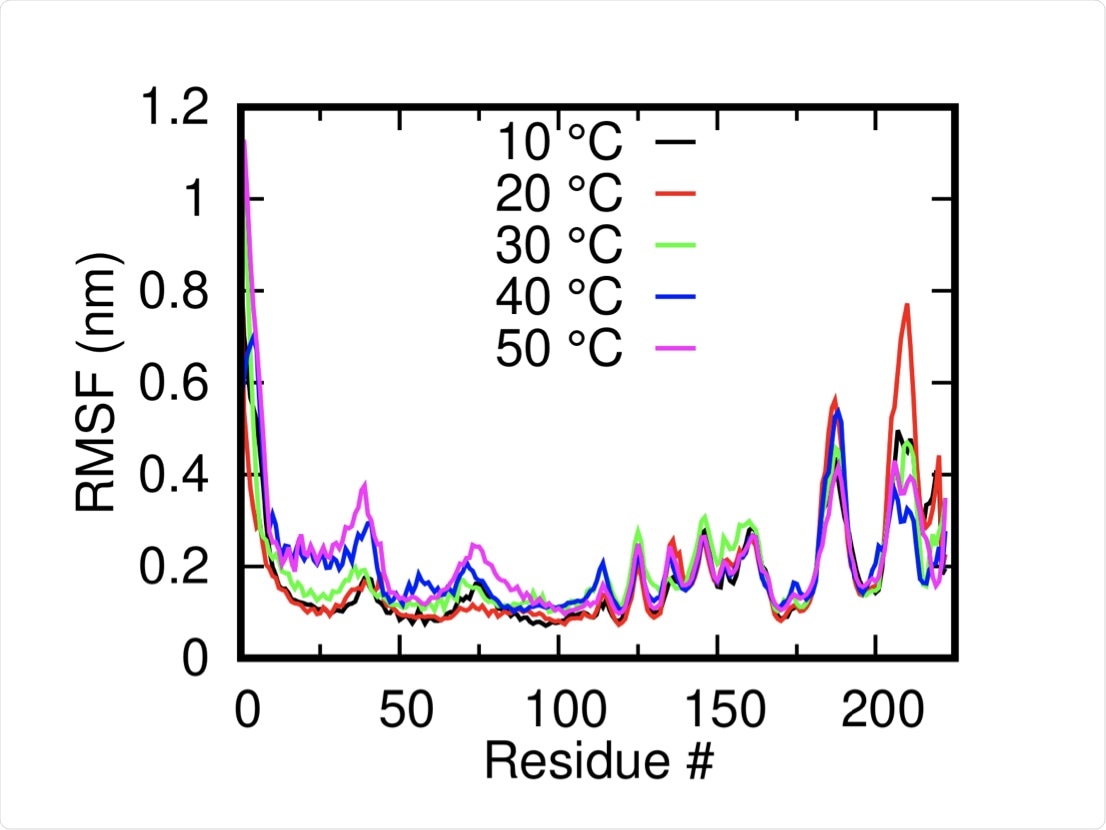 Root Mean Squared Fluctuation of the M-protein monomers (mean of both dimers) at different temperatures shows the overall stability of the systems.
Root Mean Squared Fluctuation of the M-protein monomers (mean of both dimers) at different temperatures shows the overall stability of the systems.
Next, the group simulated individual M protein dimers in free space. Two reference sites in the C- and N-terminal domains were selected to track the distance between the monomers at each temperature.
At low temperatures, the dimer enters a metastable low energy conformation that changes little from 10-30 ˚C, with the most stable conformation at the latter temperature. Comparably, a wider distribution of conformations was detected at 50 ˚C, owing to greater thermal fluctuation. A stable conformation distinct from that seen at 10-30 and 50 ˚C was noted for the proteins at 40 ˚C, with a narrow distribution and high Gibbs free energy.
The group next examined the character of the lipid membrane and found increasing fluctuation and disorder at elevated temperatures. The transition temperature of the specific DPPG:DPPG 7:3 mixture from gel to liquid crystalline states was found to be 41˚C, with a sharp increase in membrane flexibility is observed at this temperature.
To test the influence of temperature on membrane protein aggregation, 8 membrane protein dimers were next modeled on a lipid membrane simultaneously. At temperatures of 10-20 ˚C, no aggregation was observed. Comparably, at 30 ˚C, membrane protein dimer clusters began to form within 100 ns, with the process complete by 300 ns.
The rate of aggregation of membrane protein dimers is related to the degree of disorder of the lipid membrane. Thus, at 40 ˚C, aggregation initiated within the first few ns of the simulation, with steady aggregation taking place until 250 ns, and at 50 ˚C very fast aggregation was observed, with two separate clusters forming.
A larger simulation featuring 64 membrane protein dimers on a lipid bilayer was subsequently performed at 30 and 40 ˚C, with similar clustering behavior observed. Partially or fully formed clusters were noted to have an influence on membrane curvature of the lipid bilayer, thus indicating the role of the M protein in viral budding and fission events.
It is unclear from this study and others whether the M protein is solely responsible for budding events. Therefore, the authors anticipate that future work may reveal the intricacies of this relationship.
*Important notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
[ad_2]
Source link